Bula do Latisse
Princípio Ativo: Alfassebelipase
Classe Terapêutica: Outros Produtos Oftalmológico Tópicos
Latisse, para o que é indicado e para o que serve?
Latisse® é indicado para o tratamento de hipotricose palpebral (crescimento inadequado ou insuficiente de cílios).
Como o Latisse funciona?
Latisse® é uma solução de uso tópico que proporciona aumento do crescimento dos cílios em comprimento, espessura/abundância e intensidade da coloração/escurecimento. A ação do medicamento se inicia logo após o início do tratamento, porém os estudos clínicos mostram diferenças significativas de comprimento, espessura e escurecimento dos cílios que são observados a partir do 2º mês de tratamento.
Quais as contraindicações do Latisse?
Latisse® é contraindicado para pessoas que apresentam hipersensibilidade (alergia) à bimatoprosta ou a outros componentes da fórmula.
Como usar o Latisse?
Você deve usar este medicamento exclusivamente na pele da pálpebra superior, na base dos cílios (onde os cílios estão em contato com a pele).
Antes da aplicação, retire as lentes de contato, se usá-las, retire qualquer produto cosmético e lave o rosto, especialmente, a região dos olhos.
A solução já vem pronta para uso. Não encoste a ponta do frasco nos dedos e nem em outra superfície qualquer, para evitar a contaminação do frasco e da solução.
Retire os aplicadores da bandeja. Posicione o aplicador horizontalmente, coloque uma gota de Latisse® na parte do aplicador mais próxima da ponta, mas não na própria ponta. Veja a figura 1.

A dose usual é de 1 gota para cada pálpebra, uma vez ao dia, (de preferência à noite), com intervalo de aproximadamente 24 horas entre as doses. A dose não deve exceder a uma dose única diária, pois foi demonstrado que administração mais frequente não produz aumento adicional do crescimento dos cílios.
Então, imediatamente arraste cuidadosamente o aplicador sobre a pele da margem da pálpebra superior na base dos cílios (onde os cílios estão em contato com a pele), a partir da parte interna da linha dos cílios (próxima do nariz) em direção à parte lateral (na direção da orelha) – veja na figura 2.
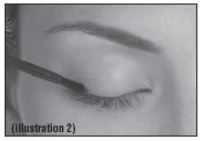
- Enxugue o excesso da solução além da margem da pálpebra.
- Descarte o aplicador usado e pegue outro aplicador para usar na pálpebra do outro olho.
- Repita os procedimentos no outro olho utilizando novo aplicador estéril. Isto ajudará a minimizar qualquer potencial para contaminação de uma pálpebra para outra.
Não aplique a solução dentro do olho, nem na pálpebra inferior. Apenas use os aplicadores estéreis fornecidos na embalagem de Latisse® para aplicar a solução.
Se a solução de Latisse® atingir o olho propriamente dito, não é esperado que isso cause danos ao olho. Retire o excesso da solução e leia atentamente as instruções de aplicação para evitar que ocorra novamente.
Feche bem o frasco depois de usar.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
O que devo fazer quando me esquecer de usar o Latisse?
Caso esqueça de aplicar o medicamento, aplique a próxima dose no dia seguinte, no horário habitual, à noite.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou do cirurgião-dentista.
Quais cuidados devo ter ao usar o Latisse?
Latisse® se destina exclusivamente para aplicação sobre a pele das margens das pálpebras superiores, nas bases dos cílios. Não deve ser aplicado na pálpebra inferior.
Contaminação da solução ou dos aplicadores de Latisse®
O frasco de Latisse® deve ser mantido intacto durante o uso. Não encoste a ponta do frasco nos olhos, nos dedos e nem em outra superfície qualquer, para evitar a contaminação do frasco e da solução. Os aplicadores estéreis incluídos na embalagem do produto devem apenas ser utilizados uma única vez (em um dos olhos) e em seguida devem ser descartados, uma vez que a reutilização do aplicador aumenta o potencial para contaminação e infecções. É importante utilizar a solução de Latisse® de acordo com as instruções, colocando uma gota da solução no aplicador de uso único por olho. Há relatos de ceratite bacteriana associada com o uso de recipientes de múltiplo uso de produtos de uso oftálmico. Estes recipientes foram inadvertidamente contaminados pelos pacientes que, na maioria dos casos, tinham uma doença ocular concomitante. Pacientes com ruptura da superfície do epitélio ocular possuem maior risco de desenvolverem ceratite bacteriana.
Efeitos sobre a pressão intraocular
Latisse® pode reduzir a pressão intraocular (PIO), principalmente se entrar em contato com o olho. Em estudos clínicos, em pacientes com ou sem PIO elevada, Latisse® reduziu a PIO; entretanto, a magnitude da redução não foi motivo de preocupação clínica. Em estudos sobre o emprego da bimatoprosta a 0,03% no tratamento da hipertensão ocular, foi demonstrado que a exposição do olho a mais do que uma dose de bimatoprosta ao dia pode diminuir o efeito redutor da pressão intraocular. Em pacientes que utilizam a bimatoprosta ou outros análogos da prostaglandina para o tratamento da pressão intraocular elevada, o uso concomitante de Latisse® pode interferir com a redução da PIO desejada.
Se você estiver utilizando outros medicamentos para reduzir a pressão intraocular elevada ou se você tem histórico de pressão ocular anormal, você deve apenas utilizar Latisse® se estiver sendo rigorosamente acompanhado por seu médico oftalmologista.
Pigmentação da pálpebra
Latisse® pode causar escurecimento da pele da pálpebra, o qual pode ser reversível. É esperado aumento da pigmentação à medida que o medicamento é utilizado, mas foi relatado que esse aumento é reversível com a descontinuação do tratamento na maioria dos pacientes.
Pigmentação da íris
Foi observado o escurecimento da íris quando Latisse® foi aplicado diretamente no olho, podendo causar um efeito permanente.
Este potencial pode ser reduzido com o uso adequado do produto (utilização dos aplicadores que acompanham o produto e dose recomendada). A alteração da pigmentação é devida ao conteúdo de melanina aumentado nos melanócitos, e não a um aumento do número de melanócitos. Os efeitos de longo prazo da pigmentação aumentada não são conhecidos. As alterações na coloração da íris observadas com a administração da bimatoprosta em solução oftálmica podem não ser notadas por vários meses a anos. Nem os nevos nem sardas da íris parecem ser afetados pelo tratamento.
Crescimento de pelos fora da área de tratamento
Existe o potencial de ocorrer crescimento de pelos em áreas onde a solução de Latisse® entra em contato repetidamente com a superfície da pele.
É importante aplicar a solução apenas na pele da margem da pálpebra superior na base dos cílios utilizando o aplicador estéril que acompanha o frasco da solução, e enxugar cuidadosamente qualquer excesso de Latisse® na margem palpebral para evitar que a solução escorra pelo rosto ou outras áreas.
Inflamação intraocular
Latisse® deve ser utilizado com cautela em pacientes com inflamação ativa intraocular (por exemplo, uveíte), pois a inflamação pode ser exacerbada.
Edema macular
A ocorrência de edema macular, incluindo edema macular cistóide, foi relatada durante o tratamento com bimatoprosta solução oftálmica a 0,03% em pacientes com pressão dos olhos aumentada. Latisse® deve ser utilizado com cautela em pacientes afácicos, em pacientes pseudoafácidos com cápsula do cristalino posterior lacerada, ou em pacientes com conhecidos fatores de risco para edema macular (por exemplo, cirurgia intraocular, oclusão da veia da retina, doença inflamatória ocular e retinopatia diabética).
Interrupção do tratamento
Se você interromper o tratamento com Latisse®, é esperado que os seus cílios voltem às condições anteriores no prazo de algumas semanas a meses. É esperado que qualquer escurecimento da pele das pálpebras desapareça após várias semanas a meses.
Uso durante a Gravidez e Lactação
Gravidez
Não foram realizados estudos adequados e controlados sobre a administração de Latisse® em mulheres grávidas. Considerando que os estudos sobre toxicidade reprodutiva em animais nem sempre são indicativos de resposta humana, Latisse® apenas deve ser utilizado em gestantes se os potenciais benefícios para a mãe justificarem os potenciais riscos para o feto.
Este medicamento não deve ser usado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Lactação
Não há dados disponíveis a respeito da excreção do Latisse® no leite humano, mas como os estudos em animais mostraram que a bimatoprosta é excretada pelo leite e muitos medicamentos também são excretados pelo leite, recomenda-se cautela na aplicação do medicamento a mulheres que amamentam.
Uso em crianças
O uso de Latisse® em crianças não foi avaliado. Este medicamento é de uso adulto – a partir de 18 anos.
Uso em idosos
Não foram observadas diferenças de eficácia e segurança entre pacientes idosos e de outros pacientes adultos.
Pacientes que utilizam lentes de contato
Latisse® não deve ser aplicado durante o uso de lentes de contato gelatinosas ou hidrofílicas, pois o cloreto de benzalcônio presente na fórmula pode ser absorvido pelas lentes e ocasionar descoloração das mesmas. As lentes de contato devem ser retiradas antes da aplicação de Latisse® e podem ser recolocadas após 15 minutos.
Pacientes com insuficiência renal ou hepática
Latisse® não foi estudado em pacientes com mau funcionamento dos rins ou do fígado e, portanto, deve ser utilizado com cautela nesses pacientes.
Interferência na capacidade de dirigir veículos e operar máquinas
Assim como outros medicamentos, caso ocorra visão borrada transitória após a aplicação, aguarde até o desaparecimento destes sintomas antes de dirigir ou operar máquinas.
Quais as reações adversas e os efeitos colaterais do Latisse?
Como acontece com qualquer medicamento, podem ocorrer reações indesejáveis com a aplicação de Latisse®.
As reações adversas oculares relatadas mais comumente com Latisse® por ordem de frequência foram:
- Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento): hiperemia (vermelhidão) conjuntival, eritema palpebral (vermelhidão das pálpebras), irritação ocular, prurido (coceira) nos olhos, escurecimento da pele ao redor dos olhos, escurecimento da cor dos olhos.
Outras reações adversas foram relatadas após a comercialização de Latisse®. Como os relatos de pós-comercialização são voluntários e de tamanho impreciso da população, não é possível estimar a frequência destas reações:
- Blefarite (inflamação das pálpebras), olho seco, secreção ocular, dor ocular, edema (inchaço) dos olhos, edema (inchaço) das pálpebras, irritação palpebral, dor palpebral, prurido (coceira) das pálpebras, sensação de corpo estranho nos olhos, hiperpigmentação da íris, aumento do lacrimejamento, alterações periorbitais e palpebrais associado com à atrofia da gordura periorbital e rigidez da pele, resultando no aprofundamento do sulco palpebral (enoftamilte) e ptose palpebral, esfoliação da pele da pálpebra e / ou área periorbital; visão borrada; hipersensibilidade (alergia) no local da aplicação; dor de cabeça; sensação de ardor palpebral, pele seca na região palpebral e/ou periocular, eritema (vermelhidão) periorbital, crescimento anormal de pelos, hordéolo (terçol), madarose (perda temporária de alguns cílios), tricorrexe (quebra dos cílios), rash (incluindo rash macular, eritematoso e pruritico, limitado à região dos cílios e periorbital), descoloração da pele (periorbital), triquíase (cílios crescem voltados para dentro, em direção ao olho).
Outras reações adversas relatadas com o uso da bimatoprosta 0,03% no uso oftálmico (quando instilado dentro do olho) foram:
- Reação muito comum (ocorre em mais de 10% dos pacientes): hiperemia (vermelhidão) ocular/conjuntival, crescimento dos cílios, prurido (coceira) dos olhos.
- Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento): conjuntivite alérgica, astenopia (vista cansada), blefarite (inflamação das pálpebras), escurecimento da pálpebra, edema (inchaço) conjuntival, secreção ocular, irritação ocular, dor ocular, alteração (escurecimento) da cor dos cílios, eritema (vermelhidão) palpebral, prurido (coceira) palpebral, aumento da pigmentação (escurecimento) da íris, aumento do lacrimejamento, secura ocular, ardor ocular, sensação de corpo estranho nos olhos, distúrbios visuais, visão borrada, fotofobia, ceratite puntacta (inflamação da córnea), hiperpigmentação (escurecimento) da pele.
- Reação incomum (ocorre entre 0,1% e 1% dos pacientes): irite (inflamação da íris), formação de crostas na margem palpebral, erosão na córnea, edema (inchaço) palpebral, meibomianite (inflamação na glândula sebácea localizada na pálpebra), opacificação e hirsutismo (crescimento aumentado de pelos).
Atenção: Todas as reações adversas observadas no uso oftálmico podem ocorrer com uso tópico, visto que existe o risco do Latisse® entrar em contato com a córnea.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
Apresentações do Latisse
Solução Tópica Estéril 0,03%
Caixa composta de frasco plástico conta-gotas contendo 5 mL de solução tópica estéril de bimatoprosta (0,3 mg/mL) acompanhado de 100 aplicadores estéreis descartáveis, de uso único.
Via de administração tópica cutânea.
Uso adulto.
Qual a composição do Latisse?
Cada mL (36 gotas) contém:
0,3 mg de bimatoprosta (0,0083 mg/gota).
Veículo: cloreto de benzalcônio, cloreto de sódio, fosfato de sódio dibásico heptaidratado, ácido cítrico monoidratado, ácido clorídrico e/ou hidróxido de sódio para ajuste de pH e água purificada.
Superdose: o que acontece se tomar uma dose do Latisse maior do que a recomendada?
Não há informações de casos de superdose em humanos. Se uma superdose ocorrer com Latisse®, o tratamento deve ser sintomático e de suporte. Nestes casos o médico deve ser consultado imediatamente.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Interação medicamentosa: quais os efeitos de tomar Latisse com outros remédios?
Não são previstas interações entre Latisse® e outros medicamentos.
Se você estiver utilizando medicamentos para reduzir a pressão aumentada dos olhos, consulte o seu médico antes de utilizar Latisse®, pois este medicamento pode alterar os efeitos desejados de medicamentos redutores da pressão intraocular.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Qual a ação da substância do Latisse?
Resultados de Eficácia
Lactentes que apresentam deficiência de LAL
O LAL-CL03 foi um estudo multicêntrico, aberto e de braço único de Alfassebelipase em 9 pacientes com deficiência de LAL com falha no crescimento ou outros indícios de doença rapidamente progressiva antes dos 6 meses de idade. Os pacientes apresentavam também doença hepática rapidamente progressiva e hepatoesplenomegalia grave. A faixa etária para admissão no estudo era de 1-6 meses. Os pacientes receberam Alfassebelipase a 0,35 mg/kg uma vez por semana durante as primeiras 2 semanas e depois 1 mg/kg uma vez por semana. Com base na resposta clínica, o aumento progressivo da dose para 3 mg/kg uma vez por semana verificou-se logo ao fim de 1 mês e até 20 meses após o início do tratamento a 1 mg/kg. Foi permitido um aumento adicional progressivo da dose para 5 mg/kg uma vez por semana. A eficácia foi avaliada comparando a experiência de sobrevida de pacientes tratados com Alfassebelipase que sobreviveram por mais de 12 meses de idade no LAL-CL03 com um grupo histórico de lactentes não tratados que apresentavam deficiência de LAL com características clínicas semelhantes.
No LAL-CL03, 6 de 9 lactentes tratados com Alfassebelipase sobreviveram mais de 12 meses (67% de sobrevivência aos 12 meses, IC 95%: 30% a 93%). Com o tratamento continuado por mais de 12 meses de idade, 1 paciente adicional faleceu aos 15 meses de idade. No grupo histórico, 0 de 21 pacientes sobreviveu mais de 8 meses de idade (0% de sobrevivência aos 12 meses, IC 95%: 0% a 16%). Alfassebelipase em doses até 1 mg/kg uma vez por semana resultou em melhorias dos níveis de alanina aminotransferase (ALT) e aspartato aminotransferase (AST) e aumento de peso nas primeiras semanas de tratamento. Da linha basal até à semana 48, as reduções médias de ALT e AST foram -34,0 U/l e -44,5 U/l, respetivamente. O aumento progressivo da dose para 3 mg/kg uma vez por semana foi associado a melhorias adicionais no aumento de peso, linfadenopatia e albumina sérica. Da linha basal até à semana 48, o percentil de peso médio para a idade melhorou de 12,74% para 29,83% e os níveis médios de albumina sérica aumentaram de 26,7 g/l para 38,7 g/l. Um lactente foi tratado com 5 mg/kg uma vez por semana no LAL-CL03; não foram notificadas reações adversas novas com esta dose. Na ausência de mais dados clínicos, esta dose não é recomendada.
Crianças e adultos com deficiência de LAL
O LAL-CL02 foi um estudo multicêntrico, duplo cego e controlado por placebo em 66 crianças e adultos com deficiência de LAL. Os pacientes foram aleatorizados para receberem Alfassebelipase a uma dose de 1 mg/kg (n=36) ou placebo (n=30) uma vez de duas em duas semanas durante 20 semanas no período duplo cego. A faixa etária no momento da randomização era dos 4 aos 58 anos de idade (71% tinham < 18 anos de idade). Para a admissão no estudo, os pacientes tinham de apresentar níveis de ALT ≥1,5 vezes o limite superior do normal (LSN). A maioria dos pacientes (58%) tinha colesterol LDL > 190 mg/dl no momento da admissão no estudo e 24% dos pacientes com colesterol LDL > 190 mg/dl estavam tomando medicamentos para baixar os lípidos. Dos 32 pacientes que fizeram uma biópsia de fígado no momento da admissão no estudo, 100% tinham fibrose e 31% tinham cirrose. A faixa etária dos pacientes com indícios de cirrose na biópsia era dos 4 aos 21 anos de idade.
Foram avaliados os seguintes parâmetros de avaliação final:
Formalização da ALT, diminuição do colesterol LDL, diminuição do colesterol não HDL, normalização da AST, diminuição dos triglicérides, aumento do colesterol HDL, diminuição do teor de gordura no fígado avaliado por imagem por ressonância magnética - eco de gradiente multi-eco (MEGE-MRI) e melhoria da esteatose hepática medida por morfometria. Observou-se uma melhoria estatisticamente significativa em vários parâmetros de avaliação final no grupo tratado com Alfassebelipase em comparação com o grupo de placebo na conclusão do período de 20 semanas de duplo cego do estudo, como apresentado na Tabela 3. A redução absoluta do nível médio de ALT foi de - 57,9 U/l (-53%) no grupo tratado com Alfassebelipase e -6,7 U/l (-6%) no grupo de placebo.
Tabela 3: Parâmetros de avaliação final primários e secundários de eficácia no LALCL02

a Proporção de pacientes que atingiram a normalização definida como 34 ou 43 U/l, em função da idade e do sexo.
b Proporção de pacientes que atingiram a normalização definida como 34-59 U/l, em função da idade e do sexo. Avaliada em pacientes com valores anormais na linha basal (n=36 para o Alfassebelipase; n=29 para o placebo).
c Avaliado em pacientes com avaliações efetuadas por MEGE-MRI (n=32 para o Alfassebelipase; n=25 para o placebo).
d Os valores de P são do teste exato de Fisher para os parâmetros de avaliação final de normalização e do teste de soma de postos Wilcoxon para todos os outros parâmetros de avaliação final.
Estiveram disponíveis biópsias de fígado emparelhadas na linha basal e na semana 20 num subgrupo de pacientes (n=26). Dos pacientes com biópsias de fígado emparelhadas, 63% (10/16) dos pacientes tratados com Alfassebelipase melhoraram da esteatose hepática (pelo menos ≥ 5% de redução) medida por morfometria em comparação com 40% (4/10) dos pacientes a receber placebo. Esta diferença não foi estatisticamente significativa. Período aberto Sessenta e cinco de 66 pacientes entraram no período aberto (até 130 semanas) com uma dose de Alfassebelipase de 1 mg/kg uma vez de duas em duas semanas.
Nos pacientes que tinham recebido Alfassebelipase durante o período de duplo cego, as reduções dos níveis de ALT durante as primeiras 20 semanas de tratamento mantiveram-se e observaram-se melhorias adicionais nos parâmetros dos lípidos incluindo os níveis de colesterol LDL e de colesterol HDL. Quatro (4) de 65 pacientes no período aberto tiveram um aumento progressivo da dose para 3 mg/kg uma vez de duas em duas semanas com base na resposta clínica. Os pacientes que receberam placebo apresentaram níveis séricos persistentemente elevados de transaminases e níveis séricos anormais de lípidos durante o período de duplo cego. Consistente com o que foi observado nos pacientes tratados com Alfassebelipase durante o período de duplo cego, o início do tratamento com Alfassebelipase durante o período aberto produziu melhorias rápidas nos níveis de ALT e nos parâmetros dos lípidos incluindo os níveis de colesterol LDL e de colesterol HDL. Num estudo aberto separado (LAL-CL01/LAL-CL04) em pacientes adultos com deficiência de LAL, as melhorias nos níveis séricos de transaminases e lípidos foram sustentadas durante o período de tratamento de 104 semanas.
População pediátrica
Cinquenta e seis de 84 pacientes (67%) que receberam Alfassebelipase durante os estudos clínicos (LAL-CL01/LAL-CL04, LAL-CL02 e LAL-CL03) pertenciam à faixa etária pediátrica e adolescente (1 mês até 18 anos de idade).
Características Farmacológicas
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Outros produtos do trato alimentar e metabolismo, enzimas; código ATC: A16AB14.
Deficiência de lipase ácida lisossomal (LAL)
A deficiência de LAL é uma doença rara associada a morbidade e mortalidade significativas, que afeta indivíduos desde a infância até à idade adulta. A deficiência de LAL nos lactentes é uma emergência médica com rápida progressão da doença ao longo de um período de semanas, tipicamente fatal nos primeiros 6 meses de vida. A deficiência de LAL é uma doença autossômica recessiva de armazenamento lisossomal caracterizada por um defeito genético que resulta numa diminuição acentuada ou perda de atividade da enzima lipase ácida lisossomal (LAL). A atividade deficiente da enzima LAL resulta no acúmulo lisossomal de ésteres do colesterol e triglicérides.
No fígado, este acúmulo conduz a hepatomegalia, teor de gordura no fígado aumentado, elevação das transaminases indicativo de lesão crônica do fígado e progressão para fibrose, cirrose e complicações de doença hepática em fase terminal. No baço, a deficiência de LAL resulta em esplenomegalia, anemia e trombocitopenia. O acúmlo de lípidos na parede do intestino conduz a má absorção e falha no crescimento. A dislipidemia é frequente com o LDL e os triglicérides elevados e o HDL baixo, associados ao teor de gordura aumentado no fígado e às elevações das transaminases. Além da doença hepática, os pacientes com deficiência de LAL têm um risco aumentado de doença cardiovascular e aterosclerose acelerada.
Mecanismo de ação
A Alfassebelipase é uma lipase ácida lisossomal humana recombinante (rhLAL). A Alfassebelipase liga-se aos recetores da superfície celular através de glicanos expressos na proteína e é subsequentemente internalizada nos lisossomas. A Alfassebelipase catalisa a hidrólise lisossomal dos ésteres do colesterol e triglicérides para colesterol livre, glicerol e ácidos graxos livres. A substituição da atividade da enzima LAL conduz a reduções do teor de gordura no fígado e das transaminases, e ativa o metabolismo dos ésteres do colesterol e triglicérides no lisossoma, conduzindo a reduções do colesterol de lipoproteínas de baixa densidade (LDL) e do colesterol de lipoproteínas não de alta densidade, triglicérides e aumentos do colesterol HDL. A melhoria do crescimento ocorre em resultado da redução de substratos no intestino.
Propriedades farmacocinéticas
Crianças e adultos
A farmacocinética da Alfassebelipase em crianças e adultos foi determinada utilizando uma análise farmacocinética da população de 65 pacientes com deficiência de LAL que receberam infusões intravenosas de Alfassebelipase a 1 mg/kg uma vez de duas em duas semanas no LAL-CL02. Vinte e quatro pacientes tinham idades compreendidas entre os 4 e os 11 anos, 23 tinham idades compreendidas entre os 12 e os 17 anos, e 18 tinham idade ≥ 18 anos (Tabela 4). Com base numa análise não compartimental de dados de adultos (LAL-CL01/LAL-CL-04), a farmacocinética da Alfassebelipase pareceu ser não linear com um aumento da exposição mais acentuado do que o proporcional à dose observado entre as doses de 1 e 3 mg/kg. Não se observou acúmulo a 1 mg/kg (uma vez por semana ou uma vez de duas em duas semanas) ou 3 mg/kg uma vez por semana.
Tabela 4: Parâmetros farmacocinéticos médios da população

* Semana 22 para os pacientes a receber placebo reinicializada para Semana 0, isto é, primeira semana de tratamento ativo.
AUCss = Área sob a curva de concentração plasmática-tempo em estado estacionário.
Cmax = Concentração máxima.
Tmax = Tempo até à concentração máxima.
CL = Depuração.
Vc = Volume central de distribuição.
T½ = Semivida.
Lactentes (< 6 meses de idade)
No LAL-CL03, a Alfassebelipase foi eliminada da circulação sistêmica com um T½ mediana de 0,1 h (intervalo: 0,1-0,2) à dose de 3 mg/kg uma vez por semana (n = 4). A diferença em exposições à Alfassebelipase entre os grupos que receberam 0,35 mg/kg e 3 mg/kg uma vez por semana foi mais do que proporcional à dose, com um aumento de 8,6 vezes da dose, resultando num aumento de 9,6 vezes da exposição para a AUC e um aumento de 10,0 vezes para a Cmax.
Linearidade/não linearidade
Com base nestes dados, a farmacocinética da Alfassebelipase pareceu ser não linear com um aumento da exposição mais acentuado do que o proporcional à dose observado entre as doses de 1 e 3 mg/kg.
Populações especiais
Durante a análise de covariáveis do modelo de farmacocinética da população para a Alfassebelipase, constatou-se que a idade, o peso corporal e o sexo não tinham uma influência significativa na CL e no Vc da Alfassebelipase. A Alfassebelipase não foi investigada em pacientes com idades compreendidas entre os 2 e os 4 anos ou em pacientes com idade igual ou superior a 65 anos. As informações sobre a farmacocinética da Alfassebelipase em grupos étnicos não caucasianos são limitadas. A Alfassebelipase é uma proteína e prevê-se que seja metabolicamente degradada através de hidrólise péptica. Consequentemente, não se prevê que a função hepática comprometida afete a farmacocinética da Alfassebelipase.
Para os pacientes com comprometimento hepático grave existe falta de dados. A eliminação renal da Alfassebelipase é considerada uma via menor para a depuração. Para os pacientes com comprometimento renal existe falta de dados. As informações sobre o impacto de anticorpos antifármaco na farmacocinética da Alfassebelipase são limitadas.
Como devo armazenar o Latisse?
Latisse® deve ser armazenado em temperatura ambiente (entre 15ºC e 30ºC) não necessitando refrigeração.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, válido por 85 dias.
Características do medicamento
Latisse® é uma solução límpida e incolor, embalada em frasco acompanhado de aplicadores estéreis descartáveis, de uso único.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais do Latisse
MS – 1.0147.0177
Farm. Resp:
Elizabeth Mesquita
CRF-SP nº 14.337
Fabricado por:
Allergan Produtos Farmacêuticos Ltda
Guarulhos - São Paulo
Indústria Brasileira
Registrado por:
Allergan Produtos Farmacêuticos Ltda
Av. Eng. Luís Carlos Berrini, 105
Torre 3 - 18º andar - Cidade Monções
São Paulo - CEP 04571-900
CNPJ: 43.426.626/0001-77
SAC
0800-14-4077
Qualidade e Tradição a Serviço da Oftalmologia.
© 2018 Allergan. Todos os direitos reservados.
Todas as marcas registradas são de propriedade de seus respectivos donos.
Venda sob prescrição médica.
Quer saber mais?
Consulta também a Bula do Alfassebelipase
O conteúdo desta bula foi extraído manualmente da bula original, sob supervisão técnica do(a) farmacêutica responsável: Karime Halmenschlager Sleiman (CRF-PR 39421). Última atualização: 12 de Junho de 2024.
Latisse 0,3mg/mL, caixa com 1 frasco com 5mL de solução de uso dermatológico + 100 aplicadores
Ofertas Recomendadas
Melhor Oferta
À partir de
R$
20,30
Informe seu CEP para ver se o produto está disponível na sua região.
Opções de Genéricos: Clique Aqui e economize
À partir de
R$
20,30
Voltar
Dorflex Max 600mg + 70mg + 100m
Ao fazer seu pedido, você concorda com a Política de Privacidade e Termos de Uso