Crixivan 400mg, caixa com 180 cápsulas gelatinosas duras
Merck Sharp & DohmeBula do Crixivan
Seu médico prescreveu Crixivan® porque você apresenta infecção pelo HIV. Crixivan® pode ajudar a reduzir suas chances de adquirir doenças associadas à infecção pelo HIV e pode ajudar a prolongar sua vida. Crixivan® também pode ajudar a diminuir a quantidade de HIV em seu corpo (denominada “carga viral”) e aumentar sua contagem de células CD4 (T). Crixivan® pode não apresentar estes efeitos em todos os pacientes.
HIV é uma doença que se inicia no sangue e que é transmitida por contato sexual ou por contato com o sangue de pessoas infectadas.
Crixivan® interfere no processo de multiplicação do vírus, fazendo com que as novas partículas virais formadas sejam incapazes de estabelecer novos ciclos de infecção.
Crixivan® (sulfato de indinavir), MSD pertence à classe de medicamentos denominados inibidores da protease. Crixivan® é ativo contra o Vírus da Imunodeficiência Humana (HIV), auxiliando na redução da infectividade e da disseminação do vírus pelo corpo.
Não há contra-indicação relativa a faixas etárias, exceto para crianças menores de 3 anos.
Informe ao médico ou cirurgião-dentista sobre o aparecimento de reações indesejáveis.
Crixivan® não deve ser administrado a pessoas que apresentem hipersensibilidade a qualquer um de seus componentes.
Crixivan® é comercializado na forma de cápsulas e deve ser administrado por via oral. A dose usual para adultos é de 800 mg (em geral administrado como 2 cápsulas de 400 mg) a cada 8 horas.
A dose para crianças e adolescentes deverá ser determinada pelo médico.
Crixivan® deve ser administrado a cada 8 horas para se obter eficácia completa.
É muito importante tomar Crixivan® exatamente conforme prescrito para assegurar a eficácia completa do produto.
Crixivan® deve ser tomado com água 1 hora antes ou 2 horas depois das refeições. Se preferir não tomar com água, Crixivan® pode ser tomado com leite desnatado, suco, café ou chá; ou uma refeição leve, como torrada com geléia ou frutas em conserva, suco e café com leite desnatado e açúcar; ou com corn flakes, leite desnatado e açúcar. Você pode se alimentar normalmente nos horários em que não for tomar Crixivan®.
A ingestão de Crixivan® com uma refeição rica em calorias, gorduras e proteínas reduz a capacidade de seu organismo absorver o medicamento, fazendo com que sua eficácia diminua.
É importante que adultos bebam, pelo menos, 1,5 litro de líquido durante o dia. É importante também que crianças e adolescentes bebam líquidos suficiente durante o dia. O médico dirá para você a quantidade de líquido que seu filho deve beber.
É importante que você tome Crixivan® exatamente conforme a prescrição médica. Não interrompa o tratamento sem primeiro falar com seu médico.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não pode ser partido ou mastigado.
Tome Crixivan® 3 vezes por dia a cada 8 horas. No entanto, se esquecer de tomar um dose, não a tome fora do horário estabelecido. Simplesmente tome a dose seguinte como de costume, isto é, no horário normal e sem duplicar a dose.
Informe ao seu médico sobre qualquer condição médica anterior ou atual, incluindo doença hepática decorrente de cirrose, problemas renais, diabetes, hemofilia, alergias ou colesterol alto e se você está tomando medicamentos redutores de colesterol conhecidos por “vastatinas”.
Informe ao seu médico se estiver grávida ou pretender engravidar.
Uso na gravidez e amamentação
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Não se sabe se Crixivan® é prejudicial ao feto quando administrado a mulheres grávidas. Se você estiver grávida, você deve tomar Crixivan® somente se o seu médico decidir que é claramente necessário.
Informe ao seu médico se estiver amamentando. O seu médico dirá para você parar de amamentar enquanto estiver tomando Crixivan®.
Uso pediátrico
Crixivan® não deve ser administrado a crianças menores que 3 anos de idade ou que não sejam capazes de engolir cápsulas.
Posso dirigir ou operar máquinas enquanto estiver tomando Crixivan®?
Não existem informações específicas que sugiram que Crixivan® afete a capacidade de dirigir ou operar máquinas. No entanto, foram relatadas vertigem e turvação da visão durante o tratamento com Crixivan®. Se você apresentar esses sintomas, deverá evitar dirigir ou operar máquinas.
Outras Considerações
Você deve saber que Crixivan® não é uma cura da infecção por HIV e que você deve continuar desenvolvendo infecções ou outras doenças associadas à doença por HIV. Portanto, você deve continuar sob os cuidados de seu médico durante o tratamento com Crixivan®.
Em alguns pacientes com infecção avançada por HIV (AIDS) e histórico de infecção oportunista, podem ocorrer sinais e sintomas de inflamação de infecções anteriores quando se inicia tratamento com combinação anti-retroviral.
Até o momento, ainda não são conhecidos os efeitos de longo prazo do tratamento com Crixivan®. O tratamento com Crixivan® ainda não demonstrou reduzir o risco de transmissão do HIV por contato sexual ou contaminação sanguínea.
Procure seu médico para mais detalhes.
Atenção: o uso incorreto pode causar resistência do vírus do HIV e falha no tratamento.
Qualquer medicamento pode apresentar efeitos adversos ou indesejáveis, denominados efeitos colaterais. Crixivan® em geral é bem tolerado.
Houve relatos de pedras nos rins e, em alguns pacientes, resultaram em problemas renais graves, incluindo insuficiência renal. Na maioria dos casos, o comprometimento renal e a insuficiência renal foram reversíveis. Avise o seu médico se você apresentar repentinamente dor nas costas importante, com ou sem sangue na urina, causada por pedras nos rins.
Alguns pacientes tratados com Crixivan® apresentaram rápida queda de contagem de hemácias (anemia hemolítica) que em alguns casos foi grave.
Alguns pacientes tratados com Crixivan® apresentaram problemas hepáticos e, raramente, insuficiência hepática.
Ocorreram diabetes e altas taxas de açúcar no sangue (hiperglicemia) em pacientes que tomaram inibidores da protease. Em alguns destes pacientes, isto resultou em cetoacidose, uma condição séria causada por falta de controle dos níveis de açúcar no sangue. Alguns pacientes tinham diabetes antes de iniciar o tratamento com inibidores da protease, outros não. Em alguns pacientes foi necessário o ajuste da dose do medicamento antidiabético. Em outros casos, foram necessários novos medicamentos antidiabéticos.
Em alguns pacientes com hemofilia, houve relato de aumento dos sangramentos.
Ocorreram dor muscular e fraqueza graves em pacientes que tomaram inibidores da protease, incluindo Crixivan®, com medicamentos redutores do colesterol denominados “vastatinas”. Avise seu médico se você apresentar dor nas costas ou fraqueza graves.
Outros efeitos adversos incluem reações alérgicas; reações cutâneas graves; fraqueza/fadiga; baixa contagem de hemácias; problemas cardíacos, incluindo infarto do miocárdio; acidente vascular cerebral; dor/inchaço abdominal; inflamação do pâncreas; inflamação dos rins; aumento de gordura em locais como pescoço, seios, abdômen e costas; diarréia; dispepsia; náusea; tontura; cefaléia; pele ressecada; alteração de coloração da pele; perda de cabelo; unha encravada (nos dedos dos pés) com ou sem infecção; cristais na urina; entorpecimento da boca; erupções; e alteração do paladar.
Seu médico possui uma lista mais completa dos efeitos adversos.
Informe ao seu médico imediatamente se ocorrerem estes ou outros sintomas incomuns. Se a condição persistir ou piorar, procure cuidados médicos.
Além disso, informe ao seu médico se apresentar quaisquer sintomas sugestivos de reação alérgica após a tomada de Crixivan®.
Crixivan® é apresentado em frascos contendo 180 cápsulas de 100 mg, 360 cápsulas de 200 mg e 180 cápsulas de 400 mg.
Uso adulto e pediátrico.
Uso oral.
As cápsulas de Crixivan® são formuladas por:
Sal de sulfato de indinavir nas concentrações de 100 mg, 200 mg e 400 mg.
Ingredientes inativos: lactose anidra e estearato de magnésio.
Os excipientes das cápsulas são: gelatina, dióxido de titânio; dióxido de silicone e lauril sulfato de sódio.
Todos os frascos contêm um dessecante.
Procure auxílio médico imediatamente caso você tome quantidade do medicamento acima do recomendado.
Sempre informe ao seu médico sobre todos os medicamentos que esteja tomando ou pretenda tomar, incluindo os obtidos sem prescrição médica (venda livre), fitoterápicos ou suplementos nutricionais.
Informe ao seu médico se estiver tomando medicamentos para reduzir o colesterol, chamados “vastatinas”.
Crixivan® pode ser tomado com vários medicamentos comumente utilizados por pessoas infectadas pelo HIV. Estes medicamentos incluem zidovudina, didanosina, lamivudina, estavudina, fluconazol, isoniazida, claritromicina, trimetoprima/sulfametoxazol e metadona.
Outros medicamentos podem ser tomados com Crixivan®, porém requerem ajuste de dose do medicamento ou de Crixivan®. Estes medicamentos incluem rifabutina, cetoconazol, itraconazol, delavirdina e efavirenz.
Informe ao seu médico se está tomando bloqueadores do canal de cálcio (medicamentos que tratam a hipertensão ou dor torácica).
Os medicamentos que não podem ser tomados com Crixivan® são:
- Terfenadina, cisaprida, astemizol, triazolam, midazolam, pimozida, rifampicina e derivados do ergot, como tartarato de ergotamina ou tartarato de ergotamina e cafeína.
Consulte seu médico antes de tomar Crixivan® com qualquer outro medicamento.
Não se recomenda tomar Crixivan® com erva de São João (Hypericum perforatum), um produto fitoterápico comercializado como suplemento nutricional, ou com produtos contendo erva de São João, pois pode ocorrer diminuição do efeito do Crixivan® ou de outros medicamentos relacionados ao HIV.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde.
Resultados de Eficácia
Estudos Clínicos
Adultos
O estudo clínico ACTG 320 foi um estudo multicêntrico, randômico e duplo-cego realizado para comparar o efeito de Indinavir (substância ativa deste medicamento) com zidovudina (ou estavudina) e lamivudina com zidovudina (ou estavudina) mais lamivudina na progressão dos sintomas da AIDS ou morte.
Os pacientes não deveriam ter recebido tratamento com inibidor da protease e lamivudina, ter recebido zidovudina e apresentar contagem de células CD4 ≤ 200 células/mm³. Nesse estudo participaram 1.156 pacientes com infecção por HIV (17% mulheres, 28% negros, 18% hispânicos, média de idade de 39 anos, tempo médio antes da terapia com zidovudina de 21 meses). O tempo médio de acompanhamento foi de 38 semanas, com o máximo de 52 semanas.
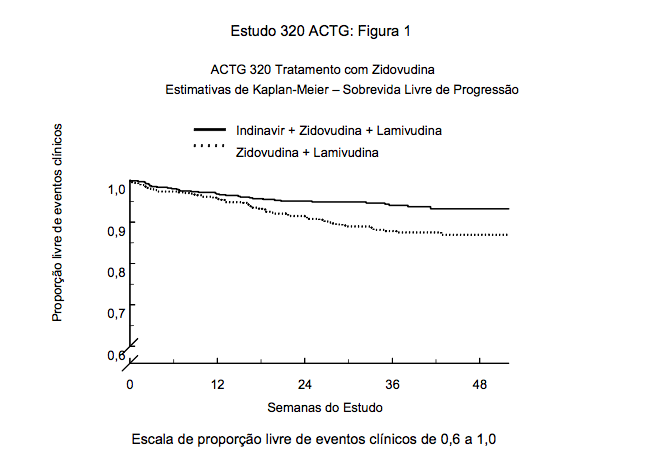
Ocorreu redução de 50% do risco na progressão dos sintomas da AIDS ou morte, em grupos tratados com uma combinação contendo Indinavir (substância ativa deste medicamento) em relação ao grupo tratado com uma combinação contendo análogo de nucleosídeo (p= 0,001). Um total de 33 (6%) dos pacientes progrediram para os sintomas da AIDS ou morte no grupo tratado com uma combinação contendo Indinavir (substância ativa deste medicamento) comparado com 63 (11%) dos pacientes do grupo tratado com combinação de análogo de nucleosídeo.
A proporção estimada de pacientes com sobrevida sem os sintomas da AIDS está apresentada na Figura 1. Além disso, ocorreu redução no risco total de mortalidade de 49% associada com Indinavir (substância ativa deste medicamento). Um total de 10 mortes (1,7%) ocorreu no grupo tratado com a combinação contendo Indinavir (substância ativa deste medicamento) e 19 (3,3%) ocorreu no grupo tratado com análogo de nucleosídeo.
A contagem média de células CD4 no período basal do estudo foi de 87 células/mm³. A alteração média na contagem de células CD4 está descrita na Figura 2.
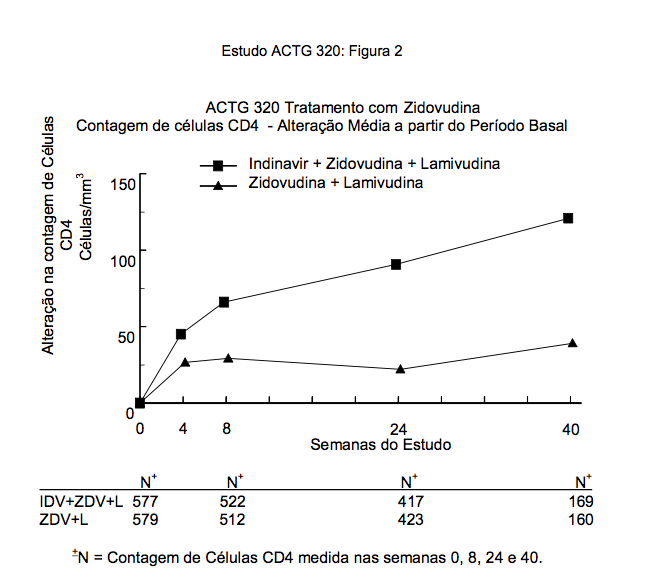
O estudo clínico 028 foi um estudo duplo-cego, multicêntrico, randômico, realizado com o objetivo de comparar os efeitos de Indinavir (substância ativa deste medicamento) mais zidovudina com os de Indinavir (substância ativa deste medicamento) isoladamente ou zidovudina isoladamente na progressão dos sintomas da AIDS ou morte e sobre as respostas de um indicador substituto.
Todos os pacientes não deveriam ter recebido tratamento anti-retroviral e apresentar contagem no número de células CD4 entre 50 e 250 células/mm³. Participaram desse estudo 996 pacientes HIV-1 soropositivos (28% mulheres, 11% negros, 1% Asiáticos/Outros, média de 33 anos de idade).
Os esquemas terapêuticos contendo zidovudina foram modificados em regime cego com adição opcional de lamivudina (o tempo médio do estudo foi de 40 semanas). O tempo médio de acompanhamento foi de 56 semanas com o máximo de 97 semanas.
Ocorreu redução no risco da progressão dos sintomas da AIDS ou morte no grupo que iniciou o tratamento com Indinavir (substância ativa deste medicamento) mais zidovudina comparado com o grupo que iniciou o tratamento com zidovudina isoladamente (p< 0,0001). Um total de 20 pacientes (6%) evoluíram com os sintomas da AIDS ou morte no grupo tratado com Indinavir (substância ativa deste medicamento) mais zidovudina comparado com 61 pacientes (18%) tratados com zidovudina isoladamente.
Ocorreu redução de 61% no risco da progressão dos sintomas da AIDS ou morte no grupo tratado com Indinavir (substância ativa deste medicamento) isoladamente comparado a um grupo tratado com zidovudina isoladamente (p< 0,0001). Um total de 26 pacientes (8%) progrediu para os sintomas da AIDS ou morte num grupo tratado com Indinavir (substância ativa deste medicamento) isoladamente.
Não existe diferença estatística significante no risco da progressão dos sintomas da AIDS ou morte entre os dois grupos que receberam Indinavir (substância ativa deste medicamento) isoladamente ou em combinação com zidovudina. A proporção estimada de pacientes que sobrevivem sem os sintomas da AIDS estão demonstrados na Figura 3.
Um total de 8 mortes (2,4%) ocorreu no grupo tratado com Indinavir (substância ativa deste medicamento) mais zidovudina, 5 (1,5%) no grupo tratado com Indinavir (substância ativa deste medicamento) isoladamente e 11 (3,3) do grupo tratado com zidovudina isoladamente.
A média da contagem das células CD4 no baseline para todos os pacientes foi de 152 células/mm³, e o RNA viral foi de 4,44 log10 cópias/ml (27,824 cópias/ml). As alterações médias na contagem CD4 e RNA viral no soro log10 são resumidos na Figura 4 e 5, respectivamente. A proporção de pacientes com RNA viral abaixo de 500 cópias/ml, o limite da quantificação da dosagem, está resumida na Figura 6.




O estudo 035, em andamento, é um estudo multicêntrico, randômico, de marcadores substitutos, para comparar os efeitos de Indinavir (substância ativa deste medicamento) com os de Indinavir (substância ativa deste medicamento) mais zidovudina, lamivudina e aqueles com zidovudina mais lamivudina na contagem de células CD4 e RNA viral.
Os pacientes não deveriam ter recebido inibidor da protease e lamivudina, ter recebido tratamento com zidovudina, apresentar contagem de células CD4 entre 50 e 400 células/mm³ e nível de RNA viral no soro ≥ 20.000 cópias/mL. Participaram desse estudo 97 pacientes soropositivos (15% mulheres, 12% latino- americanos/hispânicos, 10% negros, 4% asiáticos/outros, média de idade de 40 anos, tempo médio de terapia com zidovudina de 29,7 meses). O tratamento foi alterado para estudo aberto com Indinavir (substância ativa deste medicamento) mais zidovudina e lamivudina depois de pelo menos 24 semanas de tratamento na forma de um estudo duplo-cego e randômico.
O tempo médio de acompanhamento do estudo duplo- cego foi de 41 semanas e no máximo de 52 semanas. A contagem média de células CD4, no início do estudo (período basal), para todos os pacientes, foi de 175 células/mm³ e a média de RNA viral no soro foi de 4,62 log10 cópias/mL (41,230 cópias/mL). As alterações médias na contagem de células com RNA viral no soro em log10, durante a parte do estudo duplo-cego, estão descritas nas Figuras 7 e 8, respectivamente. A proporção de pacientes durante a fase do estudo duplo-cego RNA viral abaixo de 500 cópias/mL e o limite de quantificação da dosagem estão descritos na Figura 9.



Pacientes Pediátricos
O estudo clínico 068, em andamento, aberto, multicêntrico, para avaliar a segurança, atividade anti- retroviral e farmacocinética de indinavir na dose recomendada de Indinavir (substância ativa deste medicamento) cápsulas, 500 mg/m² a cada 8 horas, em combinação com estavudina e lamivudina em pacientes pediátricos com infecção causada pelo HIV.
Nenhum paciente do estudo deveria ter tomado inibidor da protease e nem uma dose de inibidor da transcriptase reversa, estavudina ou lamivudina. Participaram desse estudo 25 pacientes HIV-1 soropositivos (68% crianças do sexo feminino, 28% de caucasianos, 68% de negros, 4% de hispânicos) de 4 a 15 anos de idade que conseguiam engolir cápsulas. No início do estudo, a média do RNA viral no plasma foi de 4.00 log10 cópias/mL, a média da contagem de células CD4 foi de 594 células/mm³, e a porcentagem de células CD4 foi 26%. Na 24ª semana, a porcentagem de pacientes com RNA viral plasmático abaixo de 400 cópias/mL foi 60%; o qual foi inferior ao limite de quantificação no ensaio; o aumento médio na contagem de células CD4 foi 242 células/mm³, e o aumento médio percentual na contagem de células CD4 foi 4,2%.
Do estudo ACTG 395, em andamento, aberto, multicêntrico e idêntico ao projeto do Estudo 068, participaram 16 pacientes HIV-1 soropositivos (38% crianças do sexo feminino, 6% de caucasianos, 63% de negros, 31% de hispânicos) de 5 a 13 anos de idade que conseguiam engolir cápsulas. No início do estudo, a média de RNA viral plasmático foi 3,89 log10 cópias/mL, a média da contagem de células CD4 foi 678 células/mm³, e a porcentagem de células CD4 foi 30%. Na 16a semana, a porcentagem de pacientes com RNA viral plasmático abaixo de 400 cópias/mL foi 59%; o aumento médio na contagem de células CD4 foi de 73 células/mm³, e o aumento médio percentual de células CD4 foi 1,2%.
Características farmacológicas
Indinavir (substância ativa deste medicamento) é um inibidor específico da protease do vírus da imunodeficiência humana (HIV-1).
Mecanismo de ação
O indinavir inibe a protease purificada do HIV-1 e do HIV-2 com seletividade aproximadamente dez vezes maior para o HIV-1 em relação ao HIV-2. O composto liga-se diretamente ao local ativo da protease, portanto é um inibidor competitivo dessa enzima. Tal inibição impede a clivagem da poliproteína precursora viral que ocorre durante a maturação da partícula viral recém-formada.
As partículas imaturas resultantes não são infectantes e são incapazes de estabelecer novos ciclos infecciosos. O indinavir não inibe significativamente outras proteases eucarióticas, incluindo a renina, a catepsina D e a elastase humanas e o fator Xa humano.
Farmacocinética
Absorção
O indinavir foi rapidamente absorvido em jejum e o tempo decorrido para atingir o pico de concentração plasmática (Tmáx) foi de 0,8 hora (n= 11). Doses situadas entre 200–1.000 mg, administradas a indivíduos sadios e pacientes HIV-1 positivos, resultaram em aumento ligeiramente maior do que o proporcional à dose nas concentrações plasmáticas do indinavir.
Com um esquema posológico de 800 mg a cada 8 horas, a AUC (área sob a curva de concentração plasmática/tempo) em estado de equilíbrio foi de 27.813 nM•hora (n= 16) a Cmáx (pico de concentração plasmática) foi de 11.144 nM•hora (n= 16) e a concentração no vale foi de 211 nM (n= 16). No estado de equilíbrio, a concentração plasmática média do indinavir excedeu a CI95 para o HIV-1 em todos os períodos durante o intervalo posológico.
Como resultado da meia-vida curta (1,8 hora, n= 10), ocorreu apenas pequeno aumento da concentração plasmática (12%) após administrações múltiplas de 800 mg a cada 8 horas. A farmacocinética no plasma não se alterou decorridas mais de 70 semanas de administração contínua de 600 mg a cada 6 horas. A biodisponibilidade de uma dose única de 800 mg de indinavir foi de aproximadamente 65%.
Um esquema posológico com 500 mg/m² de indinavir em cápsulas, a cada oito horas, administrado a pacientes pediátricos infectados pelo HIV resultou em AUC0 – 8 h (área sobre a curva) de 27.412 nM (n= 34), Cmax de 12,182 nM (n= 34), e concentração no vale de 122 nM (n= 29). Os valores da AUC (área sobre a curva) e a Cmax foram geralmente similares àqueles observados anteriormente em pacientes adultos infectados pelo HIV que receberam a dose recomendada de 800 mg a cada 8 horas; as concentrações no vale foram mais baixas.
Efeitos dos alimentos na absorção oral
A administração do indinavir com refeições ricas em calorias, gorduras e proteínas resultou em absorção abrupta e reduzida, com aproximadamente 80% de redução da AUC (área sob a curva) e em 85% (n= 10) de redução da Cmáx.
A administração com refeições leves (por exemplo, torradas com geléia, suco de maçã, café com leite desnatado e açúcar ou sucrilhos com leite desnatado e açúcar) resultou em 2-8% de redução da AUC (área sob a curva) e da Cmáx.
A farmacocinética do indinavir ingerido como sal de sulfato de indinavir (de cápsulas abertas) misturado em papa de maçã foi geralmente comparável à farmacocinética de indinavir ingerido em cápsula em jejum.
Em pacientes pediátricos infectados pelo HIV, os parâmetros farmacocinéticos de indinavir administrados com papa de maçã foram: AUC0-8 hr (área sob a curva) de 26.980 nM•hora (n= 10); Cmax de 13,711 nM (n= 10) e concentração no vale de 146 nM (n= 9).
Distribuição
O indinavir não se liga fortemente às proteínas plasmáticas humanas (39% não se ligam). Uma rápida distribuição no tecido cerebral de rato foi demonstrada ser limitada e a razão da concentração da medicação no cérebro e no plasma calculada foi de 0,18.
A distribuição de indinavir através da barreira placentária foi significante em ratos e cachorros, mas foi limitada em coelhos. A excreção de indinavir no leite de ratas lactentes foi ampla, com a medida da razão de indinavir no leite e no plasma de 1,26 para 1,45. A distribuição dentro e fora do sistema linfático do rato demonstrou ser rápida.
Metabolismo
O metabolismo do indinavir foi avaliado em indivíduos sadios que receberam uma dose oral de 400 e 1.000 mg. Após a administração de uma dose de 400 mg de indinavir radiomarcado com 14C, aproximadamente 83% (n= 4) e 19% (n= 6) da radioatividade total foi recuperada nas fezes e na urina, respectivamente.
Foram identificados sete principais metabólitos e as conversões metabólicas foram identificadas como glucuronidação do nitrogênio piridínico, piridina-N-oxidação com e sem 3’-hidroxilação no anel indano, 3’-hidroxilação indano, p- hidroxilação da porção fenilmetil e N-despiridometilação com e sem o 3’-hidroxilação. Estudos in vitro com microssomos hepáticos humanos indicaram que o citocromo CYP34A é a única isoenzima do P450 que desempenha principal função no metabolismo oxidativo do indinavir.
Análises de amostras de plasma e urina de indivíduos que receberam indinavir indicaram que os metabólitos do indinavir contribuem pouco para a inibição da atividade da protease global in vivo.
Eliminação
Foi administrada uma dose de 200–1.000 mg para indivíduos sadios e pacientes com infecção causada pelo HIV e observou aumento da recuperação de indinavir na urina levemente maior que a dose proporcional.
O clearance renal de indinavir (116 ml/min, n= 40) é independente da faixa da dose clínica administrada. Menos de 20% do indinavir é excretado inalterado na urina. A excreção urinária média da medicação inalterada foi de 10,4% (n= 10) e 12,0% (n= 10) após a administração de uma única dose de 700 mg e 1.000 mg, respectivamente. O indinavir foi rapidamente eliminado, com uma meia-vida de 1,8 horas (n= 10).
Populações Específicas
Insuficiência hepática por cirrose
Pacientes com insuficiência hepática leve a moderada e com evidência clínica de cirrose apresentaram evidência de decréscimo do metabolismo do indinavir que resultou em AUC (área sob a curva) média aproximadamente 60% maior após uma única dose de 400 mg. A meia-vida média do indinavir aumentou para 2,8 horas, aproximadamente. Não foram estudados pacientes com insuficiência hepática grave.
Insuficiência renal
A farmacocinética do indinavir não foi estudada em pacientes com insuficiência renal. Menos de 20% do indinavir é excretado inalterado na urina.
Sexo
A farmacocinética do indinavir parece ser comparável em homens e mulheres, com base em um estudo de farmacocinética que envolveu 10 mulheres HIV positivas que receberam 800 mg Indinavir (substância ativa deste medicamento) a cada 8 horas com 200 mg de zidovudina a cada 8 horas e 150 mg de lamivudina duas vezes ao dia por uma semana.
Não existem diferenças clínicas significantes nos parâmetros farmacocinéticos comparados com homens e mulheres HIV soropositivos (dados retirados do histórico de pacientes).
Raça
A farmacocinética do indinavir não parece ser afetada pela raça.
Pacientes idosos
A segurança e a eficácia do indinavir em pacientes idosos não foi estabelecida.
Pacientes pediátricos
A farmacocinética do indinavir em adultos com infecção pelo HIV e pacientes pediátricos que receberam uma dose recomendada produziram valores de AUC e Cmáx que foram geralmente similares; para pacientes pediátricos a concentração no vale foi mais baixa.
Farmacodinâmica
Microbiologia
O indinavir na concentração de 50 a 100 nM mediou a inibição de 95% (IC95) da expansão viral (relativo ao controle de não tratados infectados pelo vírus) em culturas de células de linfócitos-T humanas infectadas com várias linhagens celulares adaptadas às variantes de HIV-1 (LAI, MN, e RF).
Observou-se inibição similar da infecção por HIV-1 em monócitos/macrófagos primários humanos, usando uma variante viral com afinidade por macrófagos (SF 162). Além disso, o indinavir na concentração de 25 a 100 nM resultou na inibição de 95% na expansão viral em culturas de células mononucleares do sangue periférico humano ativados pelo mitogênio infectados com diversos isolados clínicos primários de HIV-1, incluindo resistentes isolados para inibição de transcriptase reversa, incluindo zidovudina e inibidores da transcriptase reversa não- nucleosídeos.
Observou-se atividade anti-retroviral sinérgica quando células de linfócitos-T infectadas com a variante LAI de HIV-1 foram incubadas com indinavir junto com zidovudina, didanosina ou um inibidor da transcriptase reversa não nucleosídeo.
Resistência à medicação
A perda da supressão dos níveis de RNA viral ocorreu em alguns pacientes; entretanto, muitas vezes a contagem de células CD4 se manteve acima dos níveis anteriores ao tratamento.
Quando a perda da supressão do RNA viral ocorreu foi tipicamente associada à substituição do vírus suscetível circulante por variantes virais resistentes. A resistência foi correlacionada com o acúmulo de mutações no genoma viral que resultaram na expressão de substituições de aminoácidos na protease viral.
Foram identificadas pelo menos onze posições no resíduo de aminoácido da protease do HIV-1, nas quais as substituições foram associadas à resistência. Uma única substituição não foi capaz de provocar resistência mensurável ao inibidor; a resistência foi mediada pela co-expressão de substituições múltiplas e variáveis.
Em geral, níveis mais altos de resistência resultaram da co- expressão de números maiores de substituições nas onze posições identificadas. Substituições nessas posições pareceram se acumular seqüencialmente, provavelmente como resultado da contínua replicação viral.
Deve ser notado que a diminuição da supressão dos níveis de RNA viral foi observada mais freqüentemente quando a terapia com indinavir foi iniciada com doses mais baixas do que a dose oral recomendada de 2,4 g/dia. Portanto, a terapia com indinavir deve ser iniciada com a dose recomendada para aumentar a supressão da replicação viral e, desse modo, inibir o aparecimento de vírus resistentes.
Resistência cruzada
Isolados de pacientes infectados pelo HIV-1 com suscetibilidade reduzida ao indinavir expressaram padrões e graus de resistência cruzada variáveis a uma série de inibidores da protease, incluindo ritonavir e saquinavir.
Foi observada resistência cruzada completa entre indinavir e ritonavir; a resistência cruzada ao saquinavir, entretanto, foi variável entre os isolados. Muitas substituições de aminoácidos na protease relatadas, que foram associadas à resistência ao ritonavir e ao saquinavir, também foram associadas à resistência ao indinavir.
O uso concomitante de indinavir com um análogo de nucleosídeo pode diminuir a probabilidade de desenvolvimento de resistência ao indinavir e ao análogo de nucleosídeo.
Armazene Crixivan® na embalagem original, em temperatura abaixo de 30°C e protegido da umidade. As cápsulas de Crixivan® são sensíveis à umidade. Sempre armazene Crixivan® na embalagem original, com o dessecante.
Não tome este medicamento após a data de validade impressa na embalagem. As primeiras duas letras indicam o mês e os quatro últimos algarismos, o ano.
Não use o medicamento com o prazo de validade vencido.
Características do medicamento
Em um estudo que avaliou o sabor, a maioria dos pacientes adultos verificou que Crixivan® misturado com suco de maçã apresentava sabor desagradável. Crixivan® na forma de cápsulas, após as refeições ou em jejum, foi classificado como não apresentando sabor ou sabor melhor do que a média para um medicamento. Em geral, todas as formas foram bem-toleradas.
Não foram conduzidos estudos para avaliar o odor de Crixivan®.
- 100 mg: cápsula branca semitranslúcida, com a impressão "CRIXIVANTM 100 MG" em tinta verde.
- 200 mg: cápsula branca semitranslúcida, com a impressão "CRIXIVANTM 200 MG" em tinta azul.
- 400 mg: cápsula branca semitranslúcida, com a impressão "CRIXIVANTM 400 MG" em tinta amarela.
Antes de usar observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Registro MS - 1.0029.0004
Farmacêutico Responsável:
Fernando C. Lemos
CRF-SP nº 16.243
Produzido por:
Merck & Co., Inc., Elkton, Virginia, EUA.
Embalado por:
Merck Sharp & Dohme de México S.A. de C.V.
Av. División del Norte 3377
Colonia Xotepingo, México, D.F.
®Marca registrada de Merck & Co., Inc.,
Whitehouse Station, NJ, EUA.
MSD On Line
0800-0122232
E-mail: online@merck.com
Número de lote, data de fabricação e data de validade: vide cartucho.
Venda sob prescrição médica.
Só pode ser vendido com retenção da receita.
Especificações sobre o Crixivan
Caracteristicas Principais
Fabricante:
Tipo do Medicamento:
Novo
Necessita de Receita:
Branca Comum (Venda Sob Prescrição Médica)
Principio Ativo:
Categoria do Medicamento:
Especialidades:
Infectologia
Preço Máximo ao Consumidor:
PMC/SP R$ 1.115,26
Preço de Fábrica:
PF/SP R$ 806,73
Registro no Ministério da Saúde:
1002900040019
Código de Barras:
7897337701273
Temperatura de Armazenamento:
Temperatura ambiente
Produto Refrigerado:
Este produto não precisa ser refrigerado
Bula do Paciente:
Bula do Profissional:
Modo de Uso:
Uso oral
Pode partir:
Esta apresentação não pode ser partida
CRIXIVAN É UM MEDICAMENTO. SEU USO PODE TRAZER RISCOS. PROCURE UM MÉDICO OU UM FARMACÊUTICO. LEIA A BULA. MEDICAMENTOS PODEM CAUSAR EFEITOS INDESEJADOS. EVITE A AUTOMEDICAÇÃO: INFORME-SE COM SEU MÉDICO OU FARMACÊUTICO.




